
Within the single cell core your raw sequencing data will likely been pre-processed using the SingleCellMultiOmics package from the Alexander van Oudenaarden lab. This package holds many tools to aid with your sequencing data analysis. But with great power also comes great complexity.. As such this post will help guide you through the installation process.
Introduction
The SingleCellMultiOmics (ScMO) package has been developed within the Alexander van Oudenaarden lab by Buys de Barbanson during his doctoral research. The tools in ScMO are written mostly in the python programming language and installation will require a working python environment. However, to get the most out of this package I would recommend to install the package within a UNIX environment (Macbook/Linux). In case you only have access to a Windows computer, you can follow our other post on how to set up a Linux environment within Windows. This walk through will assume that you are able to work from a bash terminal in Ubuntu.
Installation
If this is the first time your working within a terminal in a UNIX environment the learning curve to mastering ScMO will be quite steep. But then again when was single cell sequencing analysis ever really easy? So lets just get on with it.
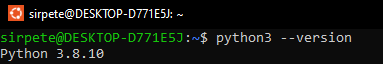
Open a “fresh” terminal window and make sure that python version older than 3.8.0 is installed by running the following command:
python3 --version
If python is not yet installed you can install python3 like this:
sudo apt install python3
Python3 is the latest version of python however there are still older python2 versions active in repositories on the internet, which in the past, have also been referenced as being called “python”. To avoid any confusion in your terminal I would strongly recommend to edit/make an alias for python to python3 in your .bashrc file.
Open the “hidden” bashrc file with the terminal text-editor nano using the following command:
nano ~/.bashrc
Use the arrows on the keyboard to navigate through the file and add the alias line to the document.

When your satisfied, press CTRL + X, to exit the editor. You will then be asked to save this data (Press Y) and finally Enter to update the current bashrc file.
restart your terminal window, and now you will be able to run python commands as if it were python3.
python --version
#Python 3.8.10
Install the singlecellmultiomics package using:
pip install singlecellmultiomics
If all goes well, the singlecellmultiomics package will install on your computer. There is however a chance that you will see a number of warnings regarding the installation location that will have to be addressed.
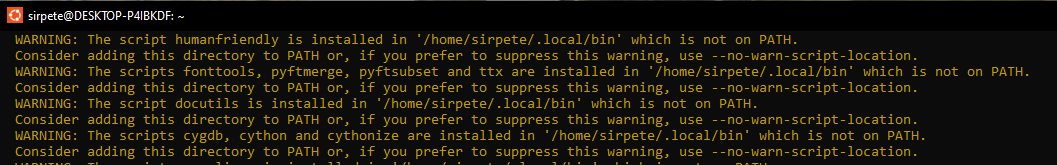
To fix this open the “hidden” bashrc file again and add the line export path (with your user name) to the document.
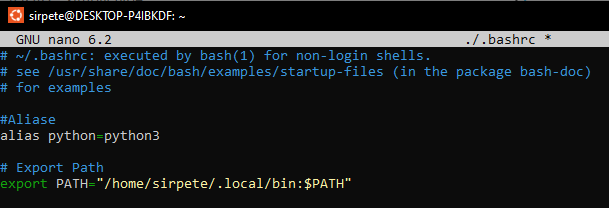
You will now have to restart your terminal, for the changes to take effect.
To make optimal use of the tools in the singlecellmultiomics package, it is highly recommended to also install the following programs:
sudo apt install bwa samtools bowtie2
(Optionally) If your intending to work on single cell transcriptome data, these tools might be useful too, but these will not be discussed here.
If this all installed smoothly, then congratulations! You are now able to work with the SingleCellMultiomics package. Curious to apply this new found power? Then check out this other blog where we dive into our pre-processing pipeline.