
It’s finally there, your sequencing data is back and you’re eager to dive in. However you quickly realize that your submitted 384 wells plate had cell groups distributed in columns but the well index is now labeled in rows.. How do you get your cell data out? Well, keep calm and please read on.
Introduction
When performing any scientific experiment there is always a need to include multiple experimental- and controls groups in order to interpret the outcome of the experiment. As a result, it is often recommended to process your experimental groups and controls together to minimize the effect any technical errors might induce. It is therefore not uncommon to include multiple cell groups on a single 384 wells plate prior to sequencing. Depending on the manner in which the cells were FACS sorted, it can happen that some data translation is required to find back of which well the data of a particular cell originated from. Surely there is a solution to this right?
Get your cells back
Whether you’ve performed scKaryo-seq or scChIC-seq all data has been pre-processed using the SingleCellMultiOmics package of the Van Oudenaarden lab. During the pre-processing stages, reads from every cell will get assigned to a corresponding cell-index. This cell-index ranges from 1 to 384, which is to be expected as there are 384 wells on the plate, one for each cell. The 384 wells plate however, are labeled as a matrix with alphabetical rows and numerical columns. 
Using R we can write a function to convert from cell-index to a well-index.
#To convert a cell-index to a well-index
index_to_well <- function(index){
wells <- NULL
for (i in toupper(letters[1:16])){
for (c in c(1:24)){
ti<-paste0(i,c)
wells <- c(wells,ti)
}
}
well_index <- wells[index]
return(well_index)
}
We can now quickly determine that cell 100 originated from well “E4”.
index_to_well(100)
#[1] "E4"
We can also write a second function to convert between well-index and cell-index.
#To convert a well-index to a cell-index
well_to_index <- function(well){
well <- toupper(well)
row.l <- substr(well,1,1)
row.nr <- substr(well,2,3)
index <- (match(row.l, toupper(letters[1:16])) -1 ) * 24
index <- index + as.integer(row.nr)
return(index)
}
Now we can supply this function with a string of a well “E4” (include the quotes!) and it will return the corresponding cell-index 100:
well_to_index("E4")
#[1] 100
Excellent, now we can easily convert back and forth. What might also be useful is to create a list of all cell names within a particular group. As an example we might want to isolate all cell names corresponding to our control group. To so we can use the previous functions to convert between indexes but it would be nice to recreate the complete cell file name, as that is how each data set is labeled. As each cell id contains the name of the 384 wells plate, a third function that inserts the plate name is required.
#Insert plate names for each well/index
insert_plate_name <- function(index, plate_name,ext=".bam"){
incl.plate_name <- NULL
for (n in 1:length(index)){
convert <- paste0(plate_name,index[n],ext)
incl.plate_name <- c(incl.plate_name, convert)
}
return(incl.plate_name)
}
This allows us to recreate a cell id from it’s constituent parts.
insert_plate_name(100, "SCC-scChIC-HUB-CORE-001_", ext="")
#[1] "SCC-scChIC-HUB-CORE-001_100"
or recreate the single cell bam file names for scKaryo-seq.
insert_plate_name(100, "sc.SCC-scKaryo-HUB-CORE-001_")
#[1] "sc.SCC-scKaryo-HUB-CORE-001_100.bam"
For our example lets assume that half of our 384 wells plate is control (pink) and the other a test group (blue).
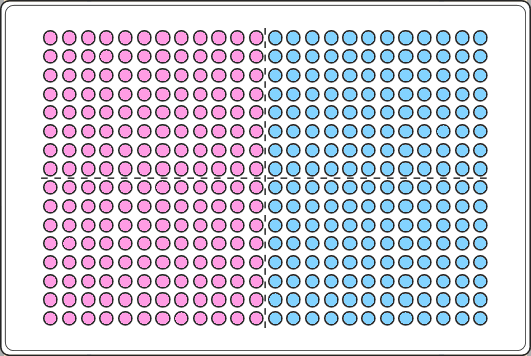
column_controls <- c(1:12) #the controls are in columns 1 to 12
row_controls <- toupper(letters[1:16]) #the control are in all rows (A until P)
###Collect all indexes from each sample
plate_controls <- NULL
for (row in row_controls){
for (c in column_controls){
t1 <- paste0(row,c)
plate_controls <- c(plate_controls,t1)
}
}
plate_controls
# [1] "A1" "A2" "A3" "A4" "A5" "A6" "A7" "A8" "A9" "A10" "A11" "A12" "B1" "B2" "B3" "B4" "B5" "B6" "B7" "B8" "B9" "B10" "B11" "B12" "C1" "C2"
# [27] "C3" "C4" "C5" "C6" "C7" "C8" "C9" "C10" "C11" "C12" "D1" "D2" "D3" "D4" "D5" "D6" "D7" "D8" "D9" "D10" "D11" "D12" "E1" "E2" "E3" "E4"
# [53] "E5" "E6" "E7" "E8" "E9" "E10" "E11" "E12" "F1" "F2" "F3" "F4" "F5" "F6" "F7" "F8" "F9" "F10" "F11" "F12" "G1" "G2" "G3" "G4" "G5" "G6"
# [79] "G7" "G8" "G9" "G10" "G11" "G12" "H1" "H2" "H3" "H4" "H5" "H6" "H7" "H8" "H9" "H10" "H11" "H12" "I1" "I2" "I3" "I4" "I5" "I6" "I7" "I8"
# [105] "I9" "I10" "I11" "I12" "J1" "J2" "J3" "J4" "J5" "J6" "J7" "J8" "J9" "J10" "J11" "J12" "K1" "K2" "K3" "K4" "K5" "K6" "K7" "K8" "K9" "K10"
# [131] "K11" "K12" "L1" "L2" "L3" "L4" "L5" "L6" "L7" "L8" "L9" "L10" "L11" "L12" "M1" "M2" "M3" "M4" "M5" "M6" "M7" "M8" "M9" "M10" "M11" "M12"
# [157] "N1" "N2" "N3" "N4" "N5" "N6" "N7" "N8" "N9" "N10" "N11" "N12" "O1" "O2" "O3" "O4" "O5" "O6" "O7" "O8" "O9" "O10" "O11" "O12" "P1" "P2"
# [183] "P3" "P4" "P5" "P6" "P7" "P8" "P9" "P10" "P11" "P12"
Now we can do the same for the experimental group.
column_test <- c(13:24) #the test cells are in columns 13 to 24
row_test <- toupper(letters[1:16]) #the test cells are in all rows (A until P)
###Collect all indexes from each sample
plate_test <- NULL
for (row in row_test){
for (c in column_test){
t1 <- paste0(row,c)
plate_test <- c(plate_test,t1)
}
}
plate_test
# [1] "A13" "A14" "A15" "A16" "A17" "A18" "A19" "A20" "A21" "A22" "A23" "A24" "B13" "B14" "B15" "B16" "B17" "B18" "B19" "B20" "B21" "B22" "B23" "B24" "C13" "C14"
# [27] "C15" "C16" "C17" "C18" "C19" "C20" "C21" "C22" "C23" "C24" "D13" "D14" "D15" "D16" "D17" "D18" "D19" "D20" "D21" "D22" "D23" "D24" "E13" "E14" "E15" "E16"
# [53] "E17" "E18" "E19" "E20" "E21" "E22" "E23" "E24" "F13" "F14" "F15" "F16" "F17" "F18" "F19" "F20" "F21" "F22" "F23" "F24" "G13" "G14" "G15" "G16" "G17" "G18"
# [79] "G19" "G20" "G21" "G22" "G23" "G24" "H13" "H14" "H15" "H16" "H17" "H18" "H19" "H20" "H21" "H22" "H23" "H24" "I13" "I14" "I15" "I16" "I17" "I18" "I19" "I20"
# [105] "I21" "I22" "I23" "I24" "J13" "J14" "J15" "J16" "J17" "J18" "J19" "J20" "J21" "J22" "J23" "J24" "K13" "K14" "K15" "K16" "K17" "K18" "K19" "K20" "K21" "K22"
# [131] "K23" "K24" "L13" "L14" "L15" "L16" "L17" "L18" "L19" "L20" "L21" "L22" "L23" "L24" "M13" "M14" "M15" "M16" "M17" "M18" "M19" "M20" "M21" "M22" "M23" "M24"
# [157] "N13" "N14" "N15" "N16" "N17" "N18" "N19" "N20" "N21" "N22" "N23" "N24" "O13" "O14" "O15" "O16" "O17" "O18" "O19" "O20" "O21" "O22" "O23" "O24" "P13" "P14"
# [183] "P15" "P16" "P17" "P18" "P19" "P20" "P21" "P22" "P23" "P24"
We now have two variables containing all the well-IDs corresponding to our control and test groups.We can convert this using our previous functions.
###convert the well IDs to indexes
index_plate_controls <- well_to_index(plate_controls)
index_plate_test <- well_to_index(plate_test)
###create a vector containing cell ids for each sample
plate_name <- "sc.SCC-scKaryo-HUB-CORE-001_"
full_plate_controls <- insert_plate_name(index_plate_controls, plate_name = plate_name, ext=".bam")
full_plate_test <- insert_plate_name(index_plate_test, plate_name = plate_name, ext=".bam")
Save the list to file.
###save the created list.
setwd("D:/work/tutorial/sc.SCC-scKaryo-HUB-CORE-001") # set this to the directory where you want to save the outputs.
write.table(full_plate_controls, file="./sc.SCC-scKaryo-HUB-CORE-001_controls.txt", sep="\n", row.names = F, col.names = F, quote = F)
write.table(full_plate_test, file="./sc.SCC-scKaryo-HUB-CORE-001_test_group.txt", sep="\n", row.names = F, col.names = F, quote = F)
With these lists at hand you can manipulate your data more easily. To get your cells out and move them to a different directory you can copy the files to a different location.
# Create copy of files
file.copy(from = paste0("D:/work/tutorial/", full_plate_controls)),
to = paste0("D:/work/experimental_controls/", full_plate_controls))
and once that is copied delete the original files.
# Remove files
#file.remove(from = paste0(""D:/work/tutorial/", full_plate_controls))
Or perhaps you’d rather combining all single cell controls into a pseudo-bulk BAM file.
samtools merge ./SCC-scKaryo-HUB-CORE-001_plate_controls_pseudo_bulk.bam -b ./plate_controls.txt
Interested in the complete R script? This can be found here.
Good luck exploring your data!